About Mucopolysaccharidosis (MPS)
Mucopolysaccharidosis (MPS) is a rare genetic disorder that results from the body’s inability to produce enzymes. Enzymes are used to break down and recycle wastes, or sugars called glycosaminoglycans (GAGS). People diagnosed with MPS lack sufficient enzymes and consequently cannot recycle GAGs. As a result, GAGs accumulate in every cell of the body – hence the name Mucopolysaccharidosis, muco meaning molecule, poly meaning many, and saccharides meaning sugars. In other words, people diagnosed with MPS have many sugar molecules in their cells. Consequently, cells do not function properly and progressive damage occurs throughout the body – such as in the heart, bones, brain, and lungs.
Mucopolysaccharidosis may not be apparent at birth because symptoms develop with age as GAGs accumulate in cells throughout the body.
Forms of MPS:
a) MPS I Hurler, Scheie, Hurler-Scheie
enzyme missing: a-L-Iduronidase
b) MPS II Hunter syndrome
enzyme missing: Iduronate sulfatase
c) MPS III A Sanfilippo, enzyme missing: Heparan N-sulfatase
MPS III B Sanfilippo, enzyme missing: a-N-Acetylglucosaminidase
MPS III C Sanfilippo, enzyme missing: Acetyl CoA: a-glycosaminide acetyltransferase
MPS III D Sanfilippo, enzyme missing: N-Acetylglucosamine 6-sulfatase
d) MPS IV A Morquio, enzyme missing: Galactose 6-sulfatase
MPS IV B Morquio, enzyme missing: B-Galactosidase
e) MPS VI Maroteaux-Lamy
enzyme missing: N-Acetylgalactosamine 4-sulfatase
f) MPS VII Sly
enzyme missing: B-Glucuronidase
g) MPS IX Hyaluronidase Deficiency (Natowicz Syndrome)
enzyme missing: Hyaluronidase
What is ML?
Mucopolipidosis (ML) is a metabolic lysosomal storage disorder in which abnormal amounts of carbohydrates and lipids (fatty substances) accumulate in cells, resulting in cell damage. ML and MPS are related diseases.
What are the Common Characteristics of MPS and Related Diseases?
Individuals diagnosed with MPS may have mental delay, cloudy corneas, short stature, stiff joints, speech and hearing impairment, heart issues, pain, and a shortened life span. However, these characteristics vary with treatment (see below).
How is MPS inherited?
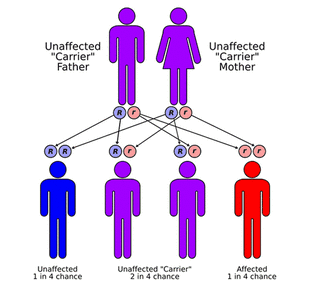
MPS and related diseases are genetic disorders. In most cases, a child receives an autosomal recessive gene from each parent, meaning that individuals are only affected if they inherit BOTH defective genes from their parents. When both parents have the defective gene, there is a 1 in 4 chance of having a child with MPS. The exception: MPS II, in which the gene is passed from a mother to her male children. Unaffected siblings may be gene carriers of the disease. MPS is a rare disorder and the occurrence of MPS in the general population is 1 in 25,000 births. However, this rate differs for each disorder. For example, MPS II Hunter syndrome only affects males and the incidence of MPS II is estimated to be 1 in 100,000-150,000 male births.
How is MPS II Hunter syndrome different?
MPS II occurs in boys because the gene associated with the disorder is located on the X chromosome, which is one of two sex chromosomes. Males only have one X chromosome and females have two. In males, one altered X chromosome can result in MPS II. In females, the mutation would have to occur in both copies of the X chromosome in order to result in MPS II. This is why MPS II is extremely rare in females - but some girls have been diagnosed with MPS II. Because fathers pass their Y chromosome and mothers pass their X chromosomes to their children, mothers are the only carriers of the condition.
If the mother is a carrier, there is a 50% risk that any boy born will have the disease. If a second boy is born without MPS, he will not be a carrier. The sisters and maternal aunts of a person with MPS II may be carriers of the disease and would also have a 50% chance of passing the syndrome to a son.
MPS II Hunter syndrome was named after Charles Hunter, a Canadian professor of medicine who was the first to describe two brothers with the disease in 1917.
Is Prenatal Diagnosis or Carrier Detection Testing Available?
Prenatal testing can be performed between 14 and 17 weeks gestation to determine if an unborn child is affected by MPS.
My sibling has MPS. Could I be a carrier?
Unaffected siblings may be gene carriers. Tests are available to determine whether individuals are carriers of an MPS or related disease. To learn more about these tests, contact your doctor, genetic center, or the National MPS Society.
Is there treatment or a cure for MPS?
Unfortunately, there is no current cure for MPS. However, there is treatment that can deliver the missing enzyme to MPS patients.
1. Enzyme Replacement Therapy (ERT) delivers enzymes to patients with MPS. ERT can reduce physical symptoms and pain. BioMarin Pharmaceutical currently produces ERT for MPS I and VI individuals. In addition, Shire Pharmaceuticals Group produced ERT treatment called Elaprase for MPS II patients.
2. Bone Marrow Transplantation (BMT) has been used to treat MPS patients but has had limited success. Physical characteristics can be improved, but neurological symptoms vary drastically. Furthermore, BMT is a risky procedure. Therefore, fewer BMTs are currently used to treat individuals affected by MPS.
Currently, ERT and BMT have limited success in treating neurological symptoms due to difficulty in delivering the enzyme across the blood brain barrier (BBB). However, there is a lot of current research and emerging therapies in the MPS field. See our news page for updates.
Examples of societies that assist MPS families:
National MPS Society (USA)
UK MPS Society
Canadian MPS Society
The Irish MPS Society
For more information about Mucopolysaccharidosis, visit the National MPS Society’s website and read their MPS and ML Fact Booklets here (http://mpssociety.org/education/mps-and-ml-booklets/)
Citation: www.mpssociety.org
Siblings with a Mission is an international organization established to serve and support siblings of individuals with special needs. All images are found on Google images and are solely used for education purposes. The stories and advice provided by Siblings with a Mission are not to be replaced by professional advice and counseling but to be considered as an additional source of support.
Mucopolysaccharidosis (MPS) is a rare genetic disorder that results from the body’s inability to produce enzymes. Enzymes are used to break down and recycle wastes, or sugars called glycosaminoglycans (GAGS). People diagnosed with MPS lack sufficient enzymes and consequently cannot recycle GAGs. As a result, GAGs accumulate in every cell of the body – hence the name Mucopolysaccharidosis, muco meaning molecule, poly meaning many, and saccharides meaning sugars. In other words, people diagnosed with MPS have many sugar molecules in their cells. Consequently, cells do not function properly and progressive damage occurs throughout the body – such as in the heart, bones, brain, and lungs.
Mucopolysaccharidosis may not be apparent at birth because symptoms develop with age as GAGs accumulate in cells throughout the body.
Forms of MPS:
a) MPS I Hurler, Scheie, Hurler-Scheie
enzyme missing: a-L-Iduronidase
b) MPS II Hunter syndrome
enzyme missing: Iduronate sulfatase
c) MPS III A Sanfilippo, enzyme missing: Heparan N-sulfatase
MPS III B Sanfilippo, enzyme missing: a-N-Acetylglucosaminidase
MPS III C Sanfilippo, enzyme missing: Acetyl CoA: a-glycosaminide acetyltransferase
MPS III D Sanfilippo, enzyme missing: N-Acetylglucosamine 6-sulfatase
d) MPS IV A Morquio, enzyme missing: Galactose 6-sulfatase
MPS IV B Morquio, enzyme missing: B-Galactosidase
e) MPS VI Maroteaux-Lamy
enzyme missing: N-Acetylgalactosamine 4-sulfatase
f) MPS VII Sly
enzyme missing: B-Glucuronidase
g) MPS IX Hyaluronidase Deficiency (Natowicz Syndrome)
enzyme missing: Hyaluronidase
What is ML?
Mucopolipidosis (ML) is a metabolic lysosomal storage disorder in which abnormal amounts of carbohydrates and lipids (fatty substances) accumulate in cells, resulting in cell damage. ML and MPS are related diseases.
What are the Common Characteristics of MPS and Related Diseases?
Individuals diagnosed with MPS may have mental delay, cloudy corneas, short stature, stiff joints, speech and hearing impairment, heart issues, pain, and a shortened life span. However, these characteristics vary with treatment (see below).
How is MPS inherited?
MPS and related diseases are genetic disorders. In most cases, a child receives an autosomal recessive gene from each parent, meaning that individuals are only affected if they inherit BOTH defective genes from their parents. When both parents have the defective gene, there is a 1 in 4 chance of having a child with MPS. The exception: MPS II, in which the gene is passed from a mother to her male children. Unaffected siblings may be gene carriers of the disease. MPS is a rare disorder and the occurrence of MPS in the general population is 1 in 25,000 births. However, this rate differs for each disorder. For example, MPS II Hunter syndrome only affects males and the incidence of MPS II is estimated to be 1 in 100,000-150,000 male births.
How is MPS II Hunter syndrome different?
MPS II occurs in boys because the gene associated with the disorder is located on the X chromosome, which is one of two sex chromosomes. Males only have one X chromosome and females have two. In males, one altered X chromosome can result in MPS II. In females, the mutation would have to occur in both copies of the X chromosome in order to result in MPS II. This is why MPS II is extremely rare in females - but some girls have been diagnosed with MPS II. Because fathers pass their Y chromosome and mothers pass their X chromosomes to their children, mothers are the only carriers of the condition.
If the mother is a carrier, there is a 50% risk that any boy born will have the disease. If a second boy is born without MPS, he will not be a carrier. The sisters and maternal aunts of a person with MPS II may be carriers of the disease and would also have a 50% chance of passing the syndrome to a son.
MPS II Hunter syndrome was named after Charles Hunter, a Canadian professor of medicine who was the first to describe two brothers with the disease in 1917.
Is Prenatal Diagnosis or Carrier Detection Testing Available?
Prenatal testing can be performed between 14 and 17 weeks gestation to determine if an unborn child is affected by MPS.
My sibling has MPS. Could I be a carrier?
Unaffected siblings may be gene carriers. Tests are available to determine whether individuals are carriers of an MPS or related disease. To learn more about these tests, contact your doctor, genetic center, or the National MPS Society.
Is there treatment or a cure for MPS?
Unfortunately, there is no current cure for MPS. However, there is treatment that can deliver the missing enzyme to MPS patients.
1. Enzyme Replacement Therapy (ERT) delivers enzymes to patients with MPS. ERT can reduce physical symptoms and pain. BioMarin Pharmaceutical currently produces ERT for MPS I and VI individuals. In addition, Shire Pharmaceuticals Group produced ERT treatment called Elaprase for MPS II patients.
2. Bone Marrow Transplantation (BMT) has been used to treat MPS patients but has had limited success. Physical characteristics can be improved, but neurological symptoms vary drastically. Furthermore, BMT is a risky procedure. Therefore, fewer BMTs are currently used to treat individuals affected by MPS.
Currently, ERT and BMT have limited success in treating neurological symptoms due to difficulty in delivering the enzyme across the blood brain barrier (BBB). However, there is a lot of current research and emerging therapies in the MPS field. See our news page for updates.
Examples of societies that assist MPS families:
National MPS Society (USA)
UK MPS Society
Canadian MPS Society
The Irish MPS Society
For more information about Mucopolysaccharidosis, visit the National MPS Society’s website and read their MPS and ML Fact Booklets here (http://mpssociety.org/education/mps-and-ml-booklets/)
Citation: www.mpssociety.org
Siblings with a Mission is an international organization established to serve and support siblings of individuals with special needs. All images are found on Google images and are solely used for education purposes. The stories and advice provided by Siblings with a Mission are not to be replaced by professional advice and counseling but to be considered as an additional source of support.
